Bringing computation to unmet needs in Human Genomics
Based in Nashville, TN, the Below lab works to improve understanding of how genetic and other molecular factors influence human health and disease.








Overview
Dr. Below’s lab develops and applies computational methods to further understanding of the genetic and epigenetic basis of human disease. Specifically, she focuses on development of novel strategies for identifying and confirming genetic risk factors to a wide range of familial and complex traits including cardiovascular and metabolic diseases, Alzheimer’s disease and other dementias, speech and language traits, oral and tooth traits, and infectious disease (pneumonia, COVID-19) via ascertainment of dense genetic, transcriptomic, and phenotypic data. She is particularly interested in the bioinformatics methods involved in network analysis of related individuals, genomic segments shared identical by descent, large-scale meta-analyses, and genetically derived predictions of expression in large electronic health record databases linked to DNA databanks.
Recent News
Waist circumference measurements along with weight and height may be a practical, low-cost marker to confirm excess body fat among children
For a long time, scientists have suspected that stuttering — a common speech condition that affects an estimated 1 in every 100 people — could be heritable. Despite how common it is, it's still a remarkably understudied condition.
A new study that identifies genetic links to stuttering may go a long way towards dampening the associated stigma.
Analysis of DNA from 23andMe users points to variants in genes linked to brain function and sense of rhythm
The Vanderbilt Genetics Institute is the intellectual hub for genomics research at Vanderbilt University Medical Center and Vanderbilt University.
Recent Publications
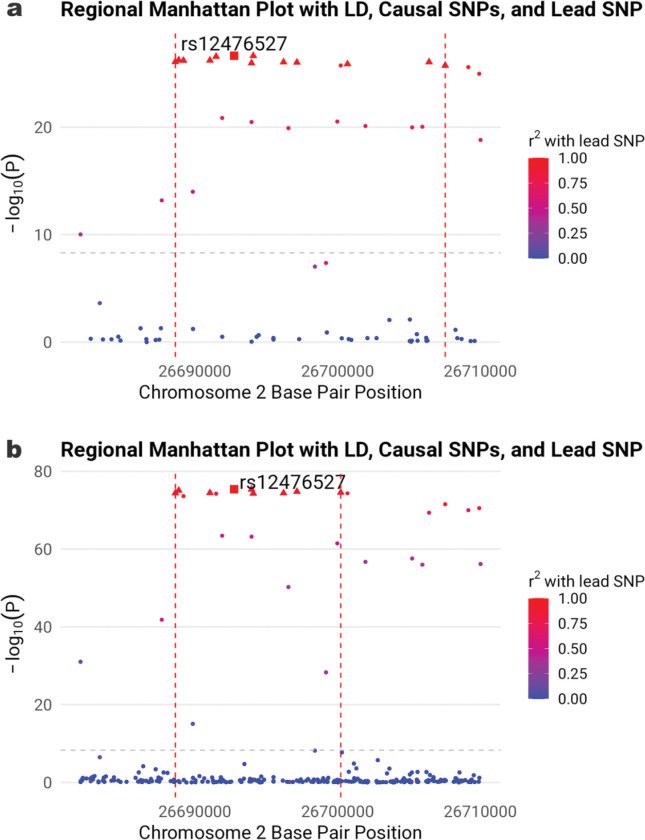
Two-degree-of-freedom fine mapping of locus on gene KCNK3
Cigarette smoking influences blood pressure (BP) levels. Studying and accounting for potential gene-smoking interactions can help discover novel loci and provide insights into biological pathways for smoking-associated BP regulation. We conducted a genome-wide association meta-analysis involving 1,188,241 individuals from 66 studies in five ancestry groups, analyzing systolic BP, diastolic BP, and pulse pressure while considering interactions between genetic variants and three smoking exposures: smoking status, cigarettes per day, and pack years. These analyses identified twelve novel loci for BP at genome-wide significance ( P<5×10−9 ), and highlighted biological processes including tight junction integrity, mitochondrial health, vascular relaxation, and endothelial function. In smoking status-stratified analyses, smoking modifies the genetic effect of six variants on BP. To prioritize likely causal, we developed and applied SuSiEgxe, a fine-mapping method based on a two-degree-of-freedom joint test using gene-environment interaction summary statistics. Fine-mapped loci uncovered immune-related pathway for smoking-associated BP regulation.
Keywords: GWAS; blood pressure; fine mapping; gene by smoking interaction; joint test.
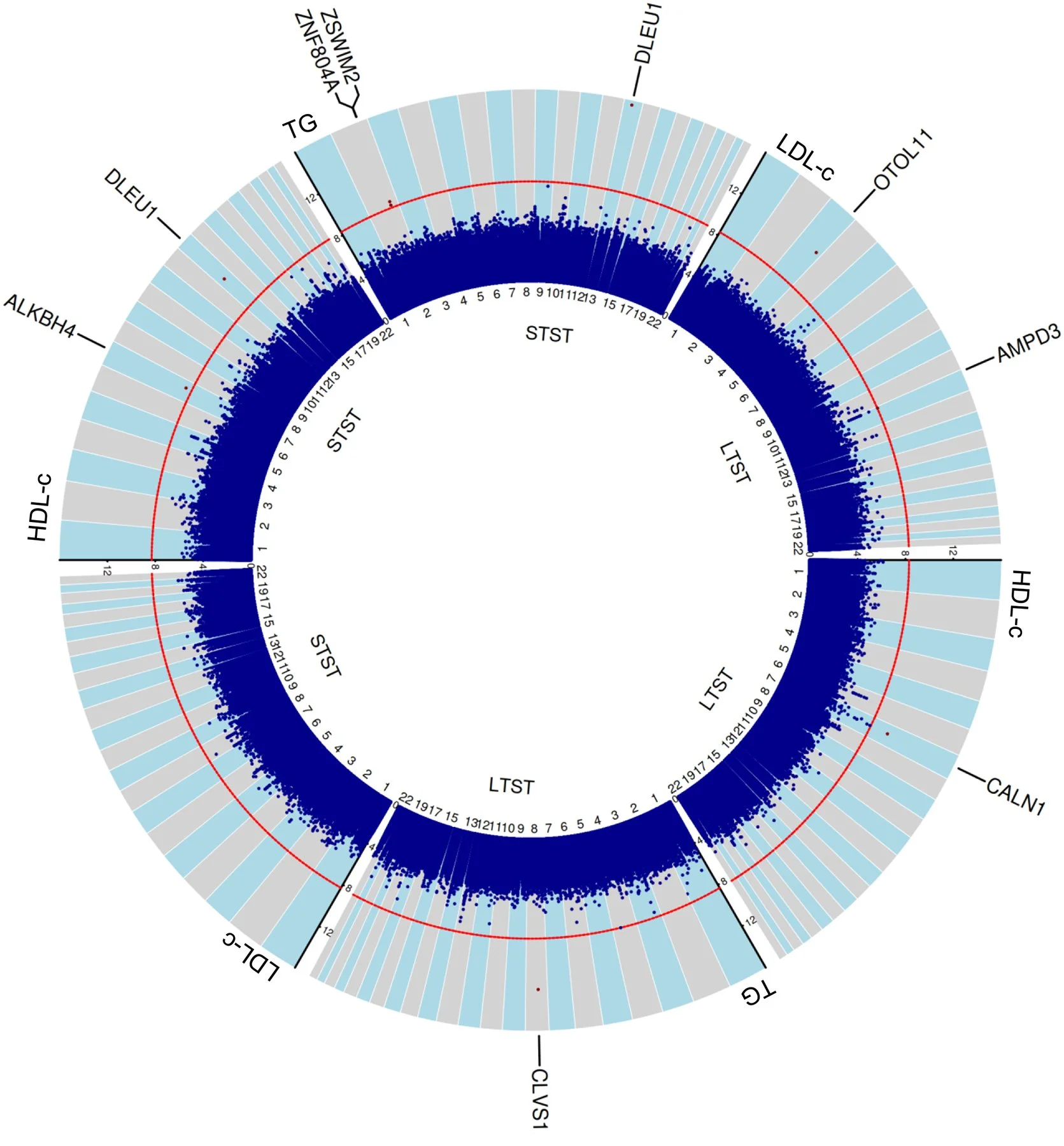
Circular -log10(Pint) plot of all the 6 main analyses
Background and aims: Deviations from the population mean in sleep duration have been associated with increased risk for developing dyslipidemia and atherosclerotic cardiovascular disease, but the mechanism of effect is poorly characterized. We performed large-scale genome-wide gene-sleep interaction analyses of lipid levels to identify genetic variants underpinning the biomolecular pathways of sleep-associated lipid disturbances and to suggest possible druggable targets.
Methods: We collected data from 55 cohorts with a combined sample size of 732,564 participants (87 % European ancestry) with data on lipid traits (high-density lipoprotein [HDL-c] and low-density lipoprotein [LDL-c] cholesterol and triglycerides [TG]). Short (STST) and long (LTST) total sleep time were defined by the extreme 20 % of the age- and sex-standardized values within each cohort. Based on cohort-level summary statistics data, we performed meta-analyses for one-degree of freedom tests of interaction and two-degree of freedom joint tests of the SNP-main and -interaction effect on lipid levels.
Results: The one-degree of freedom variant-sleep interaction test identified 10 novel loci (Pint<5.0e-9), and we additionally identify 7 loci within the two-degree of freedom analyses (Pjoint<5.0e-9 in combination with Pint<6.6e-6). Multiple loci, including those mapped to APSH (target for aspartic and succinic acid) and SLC8A1 showed biological plausibility and druggability potential based on literature.
Conclusions: Collectively, the 17 (9 with short and 8 with long sleep) loci provided evidence into the biomolecular mechanisms underlying sleep-associated lipid changes, including potential involvement of the vitamin D receptor pathway. Collectively, these findings may contribute developing novel interventions for treating dyslipidemia in people with sleep disturbances
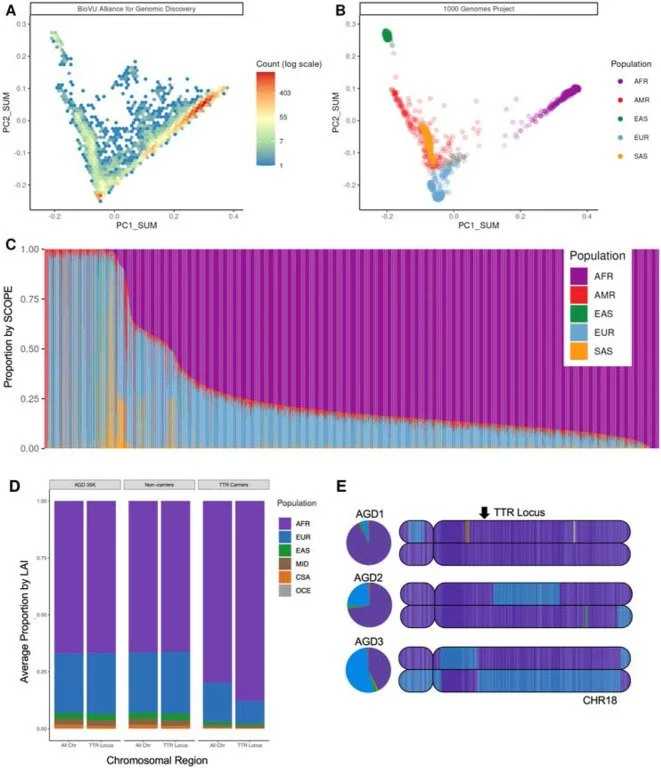
Global and local ancestry inference (LAI) in the AGD35k cohort
Genetic variation is crucial in human development, disease susceptibility, and drug response. Despite populations of African descent having the highest degree of genetic variation, genetic research has predominantly focused on populations of European descent, limiting potential for discovery. Recent studies of individuals with African ancestry have driven key medical advances benefiting all populations. Large scale, electronic health record (EHR) linked biobanks have provided opportunities to expand genetic research to larger and more diverse populations. We present initial results from the Alliance for Genomic Discovery (n = 35,024), in which 80% of participants have majority African ancestry, with genome sequencing and extensive phenotyping (median ~10 years of EHR data). We demonstrate that genetic variants known to disproportionately cause disease in patients of African descent are under-documented in the medical record, including treatable genetic conditions such as transthyretin amyloidosis. Our findings confirm that many disease-associated genetic variants have consistent effects between groups with majority African and European ancestry, including common variation, structural variation, and clonal hematopoiesis of indeterminate potential. We discover novel variants associated with drug adverse events and diagnostic codes, powered by the increased frequency of these variants in individuals with majority African ancestry. Furthermore, we report independent effects of genetic risk and social factors on glycemic control in individuals with type 2 diabetes. Overall, this work highlights the value of integrating genome sequencing and deep phenotyping in genetically diverse populations to broaden our understanding of human health and disease.